







Introduction
Fluorescent proteins are quite versatile imaging probes and have been successfully employed in virtually every biological discipline ranging from microbiology to systems physiology. These ubiquitous probes are extremely useful as reporters for gene expression studies in cultured cells, excised tissues, and whole animals. In live cells, fluorescent proteins are most commonly used to track the localization and dynamics of proteins, organelles, and other cellular compartments, while they can also be used to assess protein-protein interactions through the use of resonance energy transfer techniques (FRET ). This review provides some general tips for the practical aspects of using and imaging enhanced green fluorescent protein (EGFP) and newer members of the color palette.
For most investigators who want to track a specific protein, the application of fluorescent proteins consists of fusing the gene for the cDNA of the target gene to that of a fluorescent protein of choice followed by transferring the resulting recombinant vector into a host cell or whole organism. Pilot studies should always be undertaken with EGFP (or a high-performance derivative such as Emerald), which has been demonstrated to perform best in fusions when compared to virtually all of the other fluorescent proteins yet developed. Thus, prior to constructing vectors using fluorescent proteins in other spectral regions with coral proteins or mutated wild-type GFP protein derivatives, the integrity of the fusion engineering strategy should first be confirmed using EGFP. In the best case scenario, the new EGFP chimera and its derivative(s) from other color palette regions will enable the host protein to conduct its routine cellular duties while the piggyback fluorescent protein contributes a fluorescent molecular beacon to report on the position of the conjoined pair. Unfortunately, this doesn't always prove as straightforward as it seems, often leaving the researcher to puzzle over whether a different fluorescent protein fusion vector strategy would produce a better result.
Assuming the host cells are healthy and the transfection procedure doesn't generate too much trauma, the most common problems encountered in working with fluorescent proteins are too weak of a signal, aggregation, incorrect localization, non-functional fusions, and suppression of the expected fluorescence intensity. If these problems can be corrected, the investigator must choose between examining the cells in transient fluorescent protein transfections, where expression levels are highly varied, or taking more time to select stably-expressing cell populations that often produce superior results. It is often tempting (and much quicker) to transiently express the fluorescent protein fusion under control of a constitutive promoter and search for cells exhibiting low levels of fluorescence intensity that may correspond to expression levels minute enough as to not interfere with normal cellular function. However, producing stable cell lines presents the opportunity for a more quantitative comparison of fusion expression to that of the endogenous protein and is a much safer bet. Alternatively, placing the fusion construct into a vector having an inducible promoter enables control in modulating the level of expression. For many samples, it is possible to generate a knock-in fluorescent protein label, where the endogenous level of expression contains the fluorescent protein label. This approach is finding increased application in model organisms such as yeast, nematodes, and mice. The last approach is best for quantitative biology experiments.
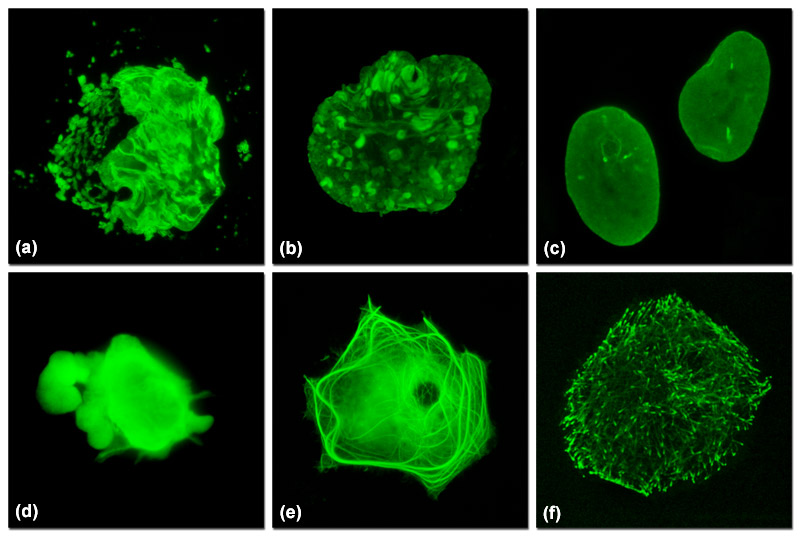
Illustrated in Figure 1 are varied fluorescent protein expression levels in transient transfections. Figures 1(a) to 1(c) visualize a fusion of mEmerald fluorescent protein to a nuclear envelope component, lamin B1, expressed at increasingly lower levels in HeLa cells. In Figure 1(a), extreme overexpression is manifested by fluorescent protein background fluorescence in the Golgi complex, aggregates in the nucleus, and distortion of nuclear envelope structures. In Figure 1(b), moderate overexpression distorts the nucleus and produces aggregates in the nucleoplasm and nuclear envelope. Finally, in Figure 1(c), low expression levels exhibit evenly distributed lamin fusion protein and distinct foci that inhabit the nucleoplasm. Similarly, Figures 1(d) to 1(f) illustrate a fusion of mEmerald with the microtubule end-binding protein, EB-3, expressed in Gray fox lung fibroblast cells. As presented in Figure 1(d), extreme overexpression pervades the cytoplasm and hampers attachment of this particular cell to the coverslip. At more moderate overexpression levels (Figure 1(e)), a fibrous network of filaments is revealed that indicates stress on microtubule function. In contrast, the low expression levels shown in Figure 1(f) clearly reveal defined EB3-labeled microtubule (+) termini, which can be followed as they migrate through the cytoplasm with time-lapse imaging. Stable transfectants should be selected from cells expressing fluorescent protein fusions at levels similar to those represented in Figures 1(c) and 1(f) for optimum results in live-cell imaging experiments.
In some cases, an EGFP fusion exhibits unexpectedly low levels of fluorescence when expressed. This problem can be due to a number of factors, including the localized environment (primarily pH) of the target organelle, improper folding of the chimera, lack of expression, or a highly unstable fusion complex. Many organelles have internal pH ranges that differ widely from that of the cytoplasm and this can interfere with fluorescent protein detection. Successful targeting of fluorescent proteins to the Golgi, endosomes, lysosomes, secretory granules, and other acidic organelles requires the use of fluorescent proteins with relatively low pKa values. For example, EGFP and Venus are more likely to experience a loss of fluorescence in the lumen of the Golgi complex than are mCherry and TagRFP, which have pKa's approximately two orders of magnitude lower. In general, fine-tuning the target environment with suitable fluorescent protein physical and biochemical parameters will ensure that intensity problems must be assigned to other variables.
Improper maturation of the fluorescent protein, the host fusion protein, or the entire chimeric complex is another source of poor fluorescence intensity, especially with some of the coral fluorescent proteins that have maturation times exceeding 12 hours or are not quite monomeric. Provided each of these entities folds properly when expressed separately, the source of error could be the amino acid linker between the fluorescent protein and its fusion protein, which should be optimized both for EGFP and the fluorescent protein under consideration. If the linker is not sufficiently long and flexible enough to separate the two domains, steric hindrance can lead to folding interference in one or both of the proteins. The first choice for linker residues is glycine, which has the smallest side chain and bestows the greatest degree of flexibility to any peptide. Interspersing several glycine residues with serine improves solubility and should be considered for longer linkers. However, the use of a wide variety of residues in fluorescent protein fusion linkers has been reported, indicating that under some circumstances there is a significant tolerance to residue choice.
In most cases, a linker length between 2 and 10 amino acids is sufficient, but the optimum size also depends upon whether the fusion occurs at the amino (N) or carboxy (C) terminus of the fluorescent protein. EGFP and other Aequorea variants have a flexible C-terminal stretch of approximately 10 amino acids, which considerably shortens the required linker length. In contrast, the N-terminal region of these probes is much less tolerant to fusions and requires a longer linker. The opposite is true for several of the coral fluorescent proteins, but the tertiary structure for many fluorescent proteins remains undetermined, so the investigator should choose linkers of at least five residues in the absence of structural data.
In cases where the fluorescent protein fusion is not expressed or targets poorly, troubleshooting efforts should be focused on the position of the fluorescent protein in fusion or the molecular integrity of the construct. If the protein is expressed but targets incorrectly, the chimera will often perform better if the fluorescent protein is placed in a different position, such as the N-terminus when the C-terminal fusion behaves poorly. Although rare, some host proteins will not tolerate a fluorescent protein fusion to either end, leaving only the choice of inserting the probe somewhere in the middle of the host sequence. The most tolerant regions are highly flexible loops, but other sequences where the amino acids are relatively disordered may work as well. Another possibility is to use circularly permuted GFP derivatives where the original amino and carboxy termini have been linked with a short spacer and new terminal ends established within the beta-barrel. A total lack of expression usually indicates a problem with the construct design (such as the fluorescent protein being inserted out of frame). After ensuring that the nucleotide sequence is correct and contains a Kozak initiation site, check to determine if the flanking sequences of the fusion gene may be interfering with transcription or translation. The remedy may be to excise the entire fusion gene and insert it into the multi-cloning site of a commercial expression vector.
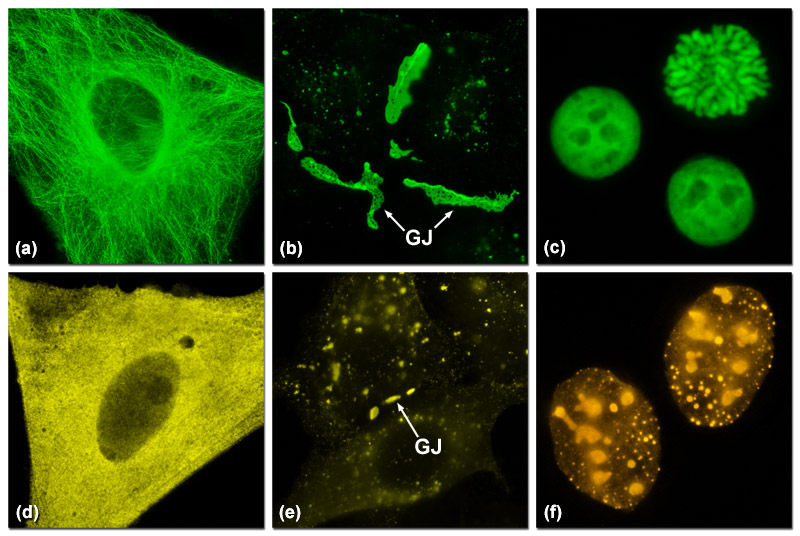
Presented in Figure 2 are several examples of good and poor localization in fluorescent protein fusions to targeting peptides and proteins. Three of the most problematic fusions are illustrated in Figure 2: alpha-tubulin, connexin Cx43, and histone H2B. Fluorescent proteins that do not display monomeric character generally perform poorly in these fusions. In Figure 2(a) a fusion of mEmerald to human alpha-tubulin expressed in HeLa cells demonstrates excellent morphology in defining the microtubule network. In contrast, Figure 2(d) shows how monomeric Kusabira Orange (mKO, a reef coral orange protein) expressed in HeLa cells displays much poorer localization than the identical mEmerald construct (punctate fluorescence with no definition of microtubules). Similarly, a fusion of gap junction forming connexin 43 fused to mEmerald forms well-defined gap junction plaques in HeLa cells (Figure 2(b)), whereas an identical fusion using mKO forms much smaller plaques that are difficult to distinguish from overlapping cell membranes (Figure 2(e)). When fused to human histone H2B, mEmerald localizes to the nucleus as expected and cells in all phases of mitosis can be imaged (Figure 2(c)). In contrast, the same fusion incorporating the tetrameric DsRed2 (Figure 2(f)) exhibits punctate labeling in the nucleus and concentrates in the nucleoli. Cells expressing this construct are rarely observed to successfully complete mitosis.
Poor localization coupled with cytoplasmic aggregation, which can occur for a wide variety of reasons, is perhaps the single largest source of problems when examining coral fluorescent protein fusion chimeras. In many cases, the primary cause is interference with the host protein's normal biological function, but aggregation and oligomerization artifacts from the fluorescent protein itself, as well as excessively high expression levels (Figure 1), can also produce the same result. In the case of coral fluorescent protein derivatives, apparent high levels of aggregation can be due to transport of the chimera to the lysosomes for degradation. This type of aggregation is not apparent with standard EGFP derivatives that have high pKa values and thus are not fluorescent in the highly acidic lysosomes. In situations where the fusion host protein itself forms a biopolymer (such as actin, tubulin, and the histones), any degree of oligomerization can be highly disruptive to proper localization and should be carefully considered before embarking on vector construction. In general, many of the newly developed monomeric orange and red proteins from coral species should be suspected of contributing artifacts if aggregation or improper localization is observed (Figure 2). It cannot be overemphasized that the best approach when using these fluorescent proteins is to compare the distribution of new fusion chimeras with the distribution of the native protein fused to a well-studied EGFP variant and to subsequently verify proper localization using immunofluorescence or synthetic dyes.
Contributing Authors
David W. Piston - Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville, Tennessee, 37232
Robert E. Campbell - Department of Chemistry, University of Alberta, Edmonton, Alberta, Canada, T6G2G2
Michael W. Davidson - National High Magnetic Field Laboratory, 1800 East Paul Dirac Dr., The Florida State University, Tallahassee, Florida, 32310.