







Introduction
Widefield and confocal fluorescence microscopy are capable of magnifying and imaging light-emitting fluorophores with a resolution that approaches a quarter of a micrometer, thus enabling the study of dynamic events and the fine structural details of cellular architecture. Using these techniques, key insights into numerous events occurring within cells, tissues, and whole organisms have been obtained, including intracytoplasmic transport of vesicles, cytoskeletal structure, mechanisms of tissue remodeling, and the migration of cancer cells in a diseased organism. Central to the recent dramatic rise in the use of fluorescence microscopy in cell biology has been the development of genetically-encoded fluorescent proteins that act as endogenous labels to enable virtually any protein or peptide to become a fluorescent homing beacon for imaging and analysis. The widespread availability of advanced new instrumentation and highly sensitive detector systems has further permitted the acquisition of superior images with spatiotemporal characteristics appropriate for addressing a diverse array of biological questions.
Despite its revolutionary impact on biology, all traditional forms of fluorescence microscopy (including widefield, confocal, and multiphoton) face a resolution limit imposed by the diffraction of light through lenses and circular apertures. The wavelike character of diffracted light prevents objects smaller than approximately 200 nanometers in the lateral (x,y) dimensions and approximately 500 nanometers in the axial (z) dimension from being visualized as anything but a blur. Due to the fact that most subcellular structures (such as actin fibers, intermediate filaments, microtubules, ribosomes, and transport vesicles) exhibit features much smaller than this size, a mechanism for breaking through the diffraction barrier, and imaging beneath the size limitation that it defines, has been the holy grail of optical microscopy for centuries.
In the past decade, several distinct conceptual strategies have been introduced to overcome the diffraction barrier and enable the analysis of biological structures at the superresolution level. One highly developed strategy, often referred to as point-spread function engineering or illumination-based superresolution, utilizes non-linear optical approaches to reduce the focal spot size. Examples of this type of superresolution imaging are stimulated emission depletion (STED), ground state depletion (GSD), and saturated structured illumination (SSIM) microscopy. By modifying the excitation light pattern through controlled engineering of the point-spread function to produce a much smaller focal spot size, these advanced techniques are capable of resolving fine structural details in biological specimens. Resolutions approaching 20 and 50 nanometers in the lateral dimensions have been achieved with STED and SSIM, respectively.
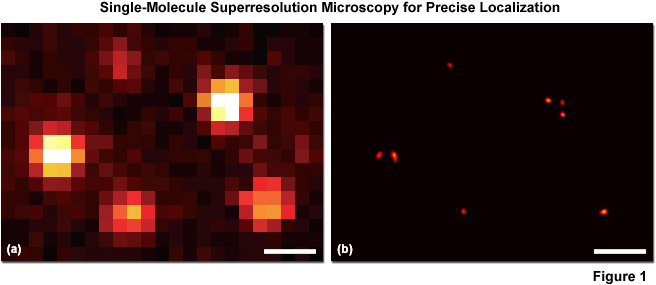
A second (and increasingly popular) strategy for overcoming the diffraction barrier employs photoswitchable fluorescent probes to resolve spatial differences in dense populations of molecules with superresolution. This approach relies on the stochastic activation of fluorescence to intermittently photoswitch individual photoactivatable molecules to a bright state, which are then imaged and photobleached. Thus, very closely spaced molecules that reside in the same diffraction-limited volume (and would otherwise be spatially indistinguishable; see Figure 1(a)), are temporally separated. Merging all of the single-molecule positions obtained by repeated cycles of photoactivation followed by imaging and bleaching produces the final superresolution image (Figure 1(b)). Techniques based on this strategy are often referred to as probe-based superresolution, and are commonly referred to by names photoactivated localization microscopy (PALM), fluorescence photoactivation localization microscopy (FPALM), and stochastic optical reconstruction microscopy (STORM). All three methods are based on the same principles, but were originally published using different photoswitchable probes. Presented in Figure 1 is an example of superresolution imaging using total internal reflection fluorescence (TIRF) microscopy. Figure 1(a) is the pixelated image of fluorescent protein single molecules attached to a coverslip. The positions of the individual molecules cannot be precisely determined because of their overlapping point-spread functions, but the center of protein molecules can be temporally resolved with high accuracy (Figure 1(b)) using PALM or similar techniques.
PALM and FPALM were developed using photoactivatable or photoconvertible fluorescent proteins (tandem dimer Eos and photoactivatable GFP, PA-GFP) as the switchable probes, whereas STORM was originally published using the synthetic carbocyanine dyes, Cy3 and Cy5, to label short DNA molecules. Regardless of the minor differences in historical development, it is possible and practical to conduct PALM experiments with synthetic dyes and STORM experiments with fluorescent proteins. In contrast to the situation with classical fluorescence microscopy, probe-based superresolution microscopy allows biological structures to be defined with nanometric accuracy, similar to the most sophisticated illumination-based superresolution techniques. However, the application of single-molecule localization imaging techniques also permits fluorescent probes highlighting subcellular structures to be individually identified at high molecular densities so that their distributions and dynamics can be analyzed. Such high resolution opens the door to many possibilities for addressing mechanistic questions regarding biological function, including the mapping of molecular machinery, as well as stoichiometry and dynamics.
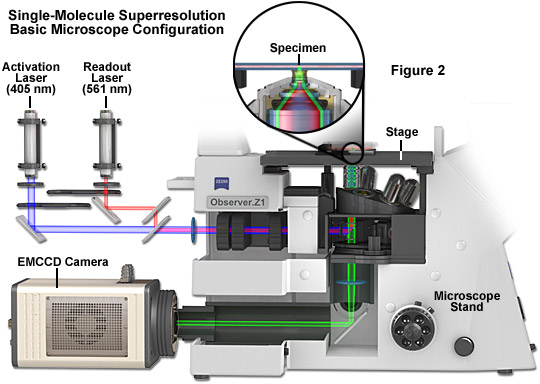
The basic microscope configuration for widefield total internal reflection (TIRF) single-molecule superresolution is presented in Figure 2. Photoactivation and readout lasers (405 and 561 nanometers, respectively) are positioned so that they can be shuttered or used simultaneously when necessary. The most versatile arrangement directly couples the lasers to the microscope without fiber optics to afford the maximum amount of power (although commercial instruments efficiently couple the laser(s) using optical fibers). A cooled electron-multiplying charge-coupled device (EMCCD) camera system is attached to the rear port of the microscope, which should receive 100 percent of the emission light for maximum signal-to-noise. The specimen is mounted in a specialized holder and bolted to the precision x-y stage for gathering transient images of single molecules. A high numerical aperture objective is positioned beneath the specimen and operates in TIRF mode. Use of an auxiliary brightfield condenser system (not shown) equipped with differential interference contrast (DIC) enables examination of the specimen to search for suitable regions of the specimen for single-molecule imaging. Central to the instrumentation is a high-speed computer system that acquires and processes images recorded by the camera.
Both illumination-based superresolution techniques, such as STED, and probe-based techniques, such as PALM, now permit biologists to visualize structures and dynamic processes in cells at or near the molecular level. The order-of-magnitude improvement in spatial resolution achieved over previous optical microscopy methodology indicates that these new approaches display enormous potential for addressing countless biological questions that require a point-to-point resolution beneath the diffraction limit of 200 nanometers. To date, superresolution microscopy has provided details on the fine architecture of cell structures such as mitochondria, lysosomes, focal adhesions, microtubules, actin filaments, and coated vesicles. Dynamic processes have also been observed, including the movement of focal adhesion complexes, sub-resolution vesicle transport, and bacteria polarity complexes, as well as single molecules constrained to the plasma membrane. No doubt, these exploratory efforts will ultimately yield to numerous investigations into the mysteries of cellular structure and function.
back to top ^Fundamental Concepts in PALM Imaging
The principle surrounding photoactivated localization microscopy and related techniques rests on a combination of imaging single fluorophores (single-molecule imaging) along with the controlled activation and sampling of sparse subsets of these labels in time. Single-molecule imaging was initially demonstrated in 1989, first at cryogenic temperatures and later at room temperature using near-field scanning optical microscopy. Since that time, the methodology has evolved to being a standard widefield microscopy technique. The prior knowledge that the diffraction-limited image of a molecule originates from a single source enables the estimation of the location (center) of that molecule with a precision well beyond that of the diffraction limit. This high level of precision is scalable with the inverse square of the number of detected photons, as will be described below.
In general terms, a single fluorescent molecule forms a diffraction-limited image having lateral and axial dimensions defined by the excitation wavelength, refractive index of the imaging medium, and the angular aperture of the microscope objective:
Resolutionx,y = λ / 2[η • sin(α)](1) Resolutionz = 2λ / [η • sin(α)]2(2)
where λ is the wavelength of light (excitation in fluorescence) and the combined term η • sin(α) is known as the objective numerical aperture (NA). Objectives commonly used in microscopy have a numerical aperture that is less than 1.5 (although new high-performance objectives closely approach this limit), restricting the term α in Equations (1) and (2) to less than 70 degrees . Therefore, the theoretical resolution limit at the shortest practical excitation wavelength (approximately 400 nanometers when using an objective having a numerical aperture of 1.40) is around 150 nanometers in the lateral dimension and approaching 400 nanometers in the axial dimension. In practical terms for imaging of enhanced green fluorescent protein (EGFP) in living cells, these values are approximately 200 and 500 nanometers, respectively. Thus, structures that lie closer than 200 to 250 nanometers cannot be resolved in the lateral plane using either a widefield or confocal fluorescence microscope.
When examining the lateral image plane in a viewfield of single molecule emitters, the central portion of each diffraction-limited spot corresponds to the probable position of a molecule and can be localized with high nanometric precision by gathering a sufficient number of photons. Methods for determining the center of localization are based on a statistical curve-fitting of the measured photon distribution (in effect, the image spot, but also referred to as the point-spread function) to a Gaussian function (see Figure 3). The ultimate goal in this exercise is to determine the center or mean value of the photon distribution (μ = x0,y0), and its uncertainty, the standard error of the mean, σ, according to the equation:
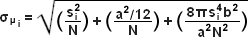 (3)
(3)
where N is the number of photons gathered, a is the pixel size of the imaging CCD detector, b is the standard deviation of the background (which includes background fluorescence emission combined with detector noise), and si is the standard deviation or width of the distribution (in direction i). The index i refers to either the x or y direction. The first term under the square root on the right-hand side of Equation (3) refers to the photon noise, whereas the second term encompasses the effect of finite pixel size of the detector. The last term takes into account the effect of background noise. By applying these simple mathematical techniques, a localization accuracy of approximately 10 nanometers can be achieved with a photon distribution of around 1000 photons when the background noise is negligible. Extension of this insight has resulted in a host of clever experiments studying isolated structures that are separated by less than the diffraction limit, such as labeled molecular motors and nanoscale DNA molecular "rulers". A major technological development in supporting probe-based superresolution is the widespread availability and ease of use of EMCCD camera systems (see Figure 2), which have single-photon sensitivity.
As described by Equation (3), the key to high-precision results for molecular localization in single-molecule superresolution imaging is to minimize background noise and maximize photon output from the fluorescent probe, a task easier described than accomplished. In the best case scenario, if 10,000 photons can be collected in the absence of background before the fluorophore bleaches or is switched off, the center of localization can be determined with an accuracy of approximately 1 to 2 nanometers. In contrast, if only 400 photons are gathered, the localization accuracy drops to around 20 nanometers or worse. Background in superresolution specimens arises from natural or transfection reagent-induced autofluorescence, as well as from residual fluorescence of surrounding probes that have entered the dark state. Thus, for probe-based single-molecule superresolution imaging techniques such as PALM, the fluorescent molecules should display a high contrast ratio (or dynamic range), which is defined as the ratio of fluorescence before and after photoactivation. Variability in contrast ratios in fluorescent proteins and synthetic fluorophores is due to differences in their spontaneous photoconversion in the absence of controlled activation.
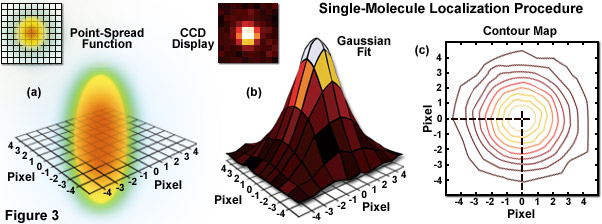
Illustrated in Figure 3 are the steps involved in localizing single molecules with high precision by fitting the point-spread function to a Gaussian function. In Figure 3(a), the point-spread function of a widefield fluorescence microscope is superimposed on a wireframe representation of the pixel array from a digital camera in both two (upper left) and three-dimensional diagrams. The pixelated point-spread function of a single fluorophore as imaged with an EMCCD is shown in the upper left of Figure 3(b), and modeled by a three-dimensional Gaussian function, with the intensity for each pixel color-mapped in the central portion of Figure 3(b). A contour map of the intensities is presented in Figure 3(c). In cases where two contour maps overlap due to emission by fluorophores with a separation distance shorter than the diffraction limit, the centroid for each fluorophore can be individually localized by subtracting the point-spread function of one fluorophore from the other (after it enters a dark state or is photobleached) due to the temporal mapping strategy for generating PALM images.
Most of the structures and organelles observed by fluorescence microscopy in biological systems are very densely populated with fluorophores, with significantly more than one molecule sharing the same diffraction-limited volume. In such cases, single-molecule localization is virtually impossible due to the fact that resulting fluorescence images appear as a highly overlapping distribution of blurred diffraction-limited spots. The mechanism around this predicament, and central to the concept surrounding PALM, is to sequentially localize sparse subsets of molecules. This concept was first demonstrated by Eric Betzig and Harald Hess a decade before PALM was introduced by resolving densely distributed luminescent centers (having distinct spectra) in a quantum well by imaging with a finely binned wavelength. As a result, although there were many centers confined within a spatial resolution volume, they became resolvable when separated along another dimension, wavelength.
The generalization of Betzig and Hess' experiment for microscopy is that if the fluorescent response from an ensemble of molecules can be spread out in some higher dimensional space, then it would be possible to image the sources individually, localize each of their centers, and then accumulate the center coordinates to create a superresolution image. The concept was successfully applied in several investigations prior to the introduction of PALM and STORM. Using laser spectroscopy, the spectra (and localization) of overlapping organic fluorophores was successfully separated, albeit at cryogenic temperatures. Subsequently, the methodology was extended to single-molecule localization through sequential photobleaching or by stochastic recruiting of fluorescent probes to a stationary binding site. Additionally, single-molecule localization has been demonstrated through the analysis of the stochastic blinking of quantum dots.
Probe-based superresolution microscopy became a reality with the discovery of photoactivatable fluorescent labels (both fluorescent proteins and synthetic fluorophores) that could be sequentially switched on in time to produce thousands of sparse subsets. The operating principle of this methodology is to start with the vast majority of fluorescent labels in the inactive state and not contributing to specimen fluorescence. A small fraction (less than 1 percent) is then activated by illumination with near-ultraviolet light (in some cases other spectral regions are useful) to induce a chemical modification in a few molecules that enables them to fluoresce. That sparse subset is then imaged and localized to generate coordinates having nanometer-level precision (see Figure 3). The registered labels are then removed by photobleaching so that a new sparse subset can be transferred into the active state and sampled to gather a new set of molecular coordinates. The process can be repeated many thousands of times up to the point that millions of such molecular coordinates are accumulated within the region being imaged. A composite image rendered from all the coordinate sets produces a single-molecule superresolution image of the fluorescently-labeled structure under investigation. Note that the molecular coordinates are not actually represented by a single point to identify their spatial position, rather a Gaussian intensity distribution corresponding to the positional uncertainty of their location is employed to build the image map.
back to top ^Molecular Localization Techniques
A host of molecular localization techniques have been described that identify more than one molecule per diffraction-limited focal spot. As discussed above, one of the first successful experiments in this field was accomplished using spectral binning on individual densely distributed luminescent species in a quantum well. A similar technique was applied to pentacene molecules embedded in a para-terphenyl crystal to yield localization precisions of approximately 40 and 100 nanometers in the lateral and axial dimensions, respectively. Another approach examined the different fluorescent lifetimes in the carbocyanine dye, Cy5, and the rhodamine derivative JF9 to localize two different molecules fluorescing with similar wavelengths within the same diffraction-limited region. Likewise, variations in photobleaching rates were used to sequentially image and localize fields of molecules until all were bleached. In this experiment, the centroid determination initiated with the most photostable molecule, localized that molecule, and repeated the process to localize up to five molecules with 10-nanometer precision in the same focal spot.
Several other approaches to high precision single-molecule localization have also been reported, but none have successfully been applied to large ensembles, such as found in a highly fluorescent focal adhesion. In contrast, PALM, and closely related techniques (such as FPALM) have extended these concepts to localizing thousands of molecules using photoconvertible or photoactivatable fluorescent proteins. The STORM technique was introduced based on the photoswitching phenomenon of common cyanine dyes when located in close proximity to each other (less than 2 nanometers). These significant advances enabled investigators to turn molecules on and off repeatedly in a controlled manner and thus maintain the requisite low density of detected molecules. Because the newly introduced single-molecule superresolution techniques of PALM, FPALM, and STORM advanced molecular localization to much higher numbers of molecules, these approaches have had a revolutionary impact on cell biology and should lead to many other new developments based on similar principles.
As mentioned, central to the concept of probe-based superresolution imaging is the ability to selectively switch a fluorescent molecule between either a bright and dark state (photoswitching) or between one fluorescence emission spectrum and another (photoconversion). This type of transition is the basis for a molecular switch, which is detailed in a concept referred to as reversible saturable/switchable optical fluorescent transition (RESOLFT). The RESOLFT concept includes switching isomerization between cis and trans states and various other optically bistable transitions in fluorophores. Virtually all reported superresolution techniques, including STED and GSD, involve some type of photoswitching or photoconversion. For example, a method referred to as PALM with independently running acquisition (PALMIRA) uses photoswitchable fluorescent proteins, whereas many other techniques, including direct STORM (dSTORM), reversible photobleaching microscopy (RPM), ground-state depletion with individual molecule return (GSDIM), and double-helix point spread function (DH-PSF) microscopy all utilize photoswitching into dark states using one or more conventional synthetic dyes, as will be discussed below.
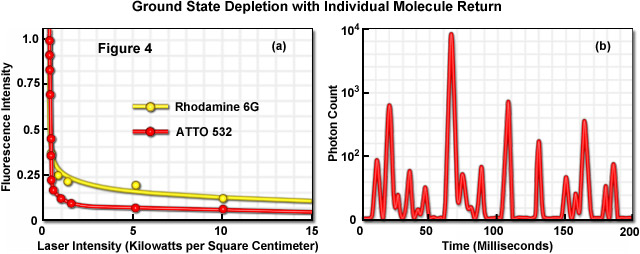
Illustrated in Figure 4 is the photoswitching of several fluorophores containing a xanthene ring system under moderate laser power to demonstrate the concept of ground state depletion and single molecule return. The graph presented in Figure 4(a) shows the rapid drop in fluorescence intensity for the probes rhodamine 6G and ATTO 532 in the presence of a triplet state quenching agent (beta-mercaptoethanol) as excitation laser intensity is increased. Note that the fluorescence intensity begins to drop rapidly at the relatively low laser power of approximately 2 kilowatts per square centimeter and then drops far more slowly (almost reaching a plateau) over the remainder of the power range. Figure 4(b) depicts a time-lapse trace of single molecule fluorescence emission from ATTO 532 molecules conjugated to goat secondary antibodies targeting mouse anti-alpha-tubulin in HeLa cells grown on coverslips. The fixed and stained cells were immersed in polyvinyl alcohol in the presence of millimolar quantities of beta-mercaptoethanol. Note that at higher laser powers, similar synthetic dyes (such as Alexa Fluor 488 and 568) will enter a long-lived dark state from which they stochastically return using only simple saline solutions without the added thiol or high-viscosity alcohol. The latter technique is referred to as reversible photobleaching microscopy, as mentioned above, although photoswitching would probably be a more appropriate term to describe the phenomenon.
back to top ^High-Density Molecular Localization
Because gathering large data sets in single-molecule localization superresolution microscopy requires extended timescales (especially compared to traditional widefield and confocal imaging), the bulk of PALM experiments are conducted on fixed cell or tissue preparations. Adherent tissue culture cells fixed with paraformaldehyde are unlikely to undergo any structural alterations so that image acquisition time is not a critical factor and is normally dictated by the number of fluorescent molecules in the specimen coupled with the patience of the investigator. Specimens in this category have produced a vast array of images revealing the molecular organization of proteins within subcellular structures at nanometer resolution, including the filamentous actin and microtubule networks. Because microtubules are distinct, well-characterized filaments having a diameter of approximately 25 nanometers and located at varying distances from each other, these structures make excellent candidates for resolution tests and microscope configuration exercises. Other potentially useful specimens for validation are focal adhesion constituents and proteins embedded within the plasma membrane or mitochondria.
A unique feature associated with all single-molecule localization techniques is that the investigator has the liberty to define the set of molecules that are displayed in the final output. For example, in some cases only molecules that localize with better than 10-nanometer uncertainty are displayed, whereas in other cases relying on the same data set, molecules that localize with 20-nanometer precision or less can be included. This versatility brings up a critical point in single-molecule PALM imaging that distinguishes between high molecular precision and the resolution that is obtainable with specific probes and specimen preparations. Even though the location of molecules within a specimen can be determined with high precision (down to a couple of nanometers in the best cases), still the stated resolution of distinct structural elements must obey the Nyquist sampling criterion of approximately two data points per resolution unit. Therefore, the density of localized molecules becomes an important factor for resolution claims, as illustrated in Figure 5 for a traditional photograph of a young boy.
The concept of localization precision, resolution, and molecular density in single-molecule localization is abstractly illustrated in Figure 5. The series of images of a young boy demonstrates how the superresolution image is constructed by plotting localized molecules, depicted in Figure 5 as points. For the purposes of illustration, the point sizes in panels (a) through (c) are depicted much larger than would be necessary to achieve the final image presented in panel (d). The boy's image in Figure 5(a) shows many localized points, but the relatively low density does not enable recognition of the image. As more points are added to the images in Figure 5(b) and 5(c), the boy becomes more recognizable, but distinguishing (resolving) the fine features associated with his face requires a higher density of data points, as shown in Figure 5(d). The important concept to note is that an image can be generated that contains precisely localized data points using single-molecule localization techniques, but the density of those points directly affects the capability to resolve features in the image.

In PALM and other single-molecule localization methods, images can be rendered with the molecules localized to the highest precisions (a nanometer or two) by selectively excluding data points with poor localization, but this success comes at the expense of the molecular density (and thus, resolution) in the final image. This situation can arise based simply on the type of specimen and target molecule of interest rather than an arbitrarily imposed limitation on molecular precision. Thus, low molecular density can arise due to the fact that the target molecule is sparsely represented in the specimen. As an example, if the average diameter of a fluorescent protein is taken to be approximately 2.5 nanometers and the protein is embedded in an organelle confined to a two-dimensional plane (such as a membrane), the maximum density of fluorescent protein molecules would be approximately 20,000 per square micrometer. Furthermore, to reach such a high density, this calculation assumes that only fluorescent protein molecules are located within this plane.
Although rare situations may arise where molecular densities are exceedingly high in biological specimens, such as the packed membrane discussed above, it is generally an unlikely physiological situation elsewhere in the cell. If fluorescent protein fusions comprise one percent of the molecules in an organelle membrane, this leads to approximately 2000 molecules per square micrometer or around two molecules every 40 to 50 nanometers. Sampling densities of this level pale in comparison to the obtainable molecular precision observed by electron microscopy and limit any resolution claims about structural features. Regardless, a PALM experiment that observes 2000 molecules per square centimeter in an organelle membrane offers a significant amount of information, despite limits on resolution. In effect, images at this density can reveal relative molecular distributions within the specimen, which is often the goal of an imaging experiment. If this data is placed into context with other known features of the specimen (such as using correlative electron microscopy), the potential increases to obtain far more information.
back to top ^Probes for Single-Molecule Superresolution Microscopy
Several prominent fluorophore classes have emerged as the best candidates for labeling subcellular targets in PALM and related techniques. These include genetically-encoded fluorescent protein fusions, synthetic dyes, quantum dots, and hybrid systems that combine a genetically-encoded target peptide with a separate synthetic component that is membrane permeant (such as the FlAsH/ReAsH system). Each specific class of fluorescent probes has its particular strengths and weaknesses, however no single class or individual fluorophore has yet been developed that combines all the preferred characteristics of an ideal probe for any application in single-molecule superresolution microscopy. The fundamental consideration for developing superresolution probes is that they must be capable of being either photoactivated, photoswitched, or photoconverted by light of a defined wavelength band as a means to alter their spectral properties for the detection of selected subpopulations.
Among the most desirable attributes for singe-molecule superresolution probes are very high brightness and contrast levels, which are necessary to maximize the number of photons that can be detected per molecule before it photobleaches or reverts to a dark, non-fluorescent state. Brightness is determined by the product of the molar extinction coefficient (εabs) and the fluorescence quantum yield (φ). Thus, the best probes have high extinction coefficients and quantum yields and provide excellent contrast over the background. In general, the brightness levels of popular synthetic dyes, such as Cy5, ATTO 650, and Alexa Fluor 488, are very high due to extinction coefficients averaging 100,000 and quantum yields exceeding 0.90. Likewise, quantum dots have equally high brightness levels. In contrast, the optical properties for optical highlighter fluorescent proteins are much less optimal with extinction coefficients ranging from approximately 15,000 to 85,000 and quantum yields between 0.05 and 0.80. Clearly, there is a significant need to genetically engineer more useful fluorescent proteins for superresolution imaging.
In addition to having high brightness levels, the best probes for superresolution microscopy should exhibit spectral profiles for the active and inactive species that are sufficiently well separated and thermally stable so that spontaneous interconversion energies are very low compared with the light-controlled activation energy. Ideally, these probes should also exhibit high switching reliability, low fatigue rates (in effect, the number of survivable switching cycles), and switching kinetics that can be readily controlled. In terms of photobleaching or photoswitching to a dark state, the best probes are those whose inactivation can be balanced with the activation rate to ensure that only a small population of molecules is activated (to be fluorescent) for readout, and that these activated molecules are separated by a distance greater than the resolution limits of the camera system. Furthermore, each photoactivated molecule should emit enough photons while in an activated state to accurately determine their lateral position coordinates.
Besides displaying the necessary fluorescence emission and other photophysical properties, PALM superresolution probes must also be capable of localizing to their intended targets with high precision and exhibit the lowest possible background noise levels. Fluorescent proteins, hybrid systems, and highly specific synthetic fluorophores (such as MitoTrackers) are able to selectively target protein assemblies or organelles, but most of the cadre of synthetic dyes and quantum dots must first be conjugated to a carrier molecule for precise labeling. In many cases, the exact proximity of the fluorescent probe to the target is questionable, as is the number of actual fluorophore units involved, especially when small synthetic dye molecules or quantum dots are conjugated to large antibodies. Also, variations in photophysical properties (such as fluorescence quantum yield) induced by environmental fluctuations or intermolecular interactions can complicate data analysis. Finally, regardless of whether localization analysis is performed on fixed or living cells, autofluorescence arising from fixatives and transfection reagents can often produce excessively high background signal, thus reducing the localization accuracy.
Properties of Selected Fluorescent Probes for Superresolution Microscopy
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 1
A compilation of properties of the most useful fluorescent proteins and synthetic dyes for superresolution microscopy is presented in Table 1. Along with the common name and/or acronym for each fluorophore, the peak excitation (Ex) and emission (Em) wavelengths, molar extinction coefficient (EC), quantum yield (QY), relative brightness, number of photons emitted per molecule (N Photons), and physiologically relevant quaternary structure are listed. C-Rhodamine and C-Fluorescein refer to caged derivatives. The computed brightness values were derived from the product of the molar extinction coefficient and quantum yield, divided by the value for EGFP. The designation ND indicates the values have not been determined, whereas NA means that the value is not applicable for the listed probe. This listing was created from scientific and commercial literature resources and is not intended to be comprehensive. The excitation and emission peak values listed may vary in published reports due in some cases to the broad spectral profiles. In actual fluorescence microscopy investigations, the experimental brightness of a particular fluorophore may differ (in relative terms) from the brightness provided in this table. Among the many potential reasons for these differences are wavelength-dependent variations in the transmission or reflectance of microscope optics and the efficiency of the camera.
The intrinsic ability of a subset of fluorescent proteins to alter their spectral properties upon exposure to light of a specific wavelength coupled with their excellent targeting specificity has been widely exploited in superresolution imaging (see the probes listed in Table 1). Although fluorescent proteins are known to undergo a variety of light-induced photoswitching characteristics, including the generation of distinct emissive and non-emissive states as well as on-and-off blinking behavior, the most useful properties are photoactivation, photoconversion, and photoswitching, properties that can be collectively termed optical highlighting (as described below). Photoactivatable fluorescent proteins are capable of being activated from a dark state to a bright fluorescent state upon illumination with ultraviolet or violet light, whereas photoconvertible fluorescent proteins can be optically transformed from one fluorescence emission bandwidth to another. Among the most useful members in the toolbox of the photoactivatable fluorescent proteins for superresolution imaging are PA-GFP and PA-mCherry1. photoconvertible fluorescent proteins that have found utility in PALM and related techniques are tandem dimer Eos (tdEos), mEos2, Dronpa, rsCherry (and the reverse derivative), and PS-CFP2.
Members of the potentially most useful class of fluorescent protein optical highlighters, those that are capable of photoswitching between a brightly fluorescent and dark state, have not been particular useful in single-molecule superresolution investigations due to low photon output in the bright state. Dronpa was used in dual-color PALM imaging, as will be subsequently discussed, but is marginal when compared to most of the photoactivatable and photoconvertible fluorescent proteins in terms of photon output per molecule. However, Dronpa features very high photoswitching contrast and emits fluorescence for an extended period, which can be advantageous for live-cell imaging (for example, using single-particle tracking as described below). Aside from the requirement that fluorescent proteins must display some type of optical highlighting behavior for superresolution imaging, they must also have sufficient brightness, chromophore maturation rates, and monomeric character to express the fusion without artifacts, such as poor targeting and dysfunction. In addition, oligomerization in fluorescent proteins can present a problem in stochastic superresolution microscopy as there is more than one chromophore per each localized probe molecule.
A rapidly growing number of synthetic organic fluorophores are emerging as excellent candidates for single-molecule superresolution imaging, including many of the traditional probes that have been used for several decades in preparing specimens for widefield and confocal fluorescence microscopy (see Table 1). Many compounds that were originally thought to be photostable have since been demonstrated to enter dark states under low oxygen conditions in the presence of aliphatic thiols. Therefore, reversible photoswitching is probably a more widespread phenomenon than was originally suspected, which opens the door to an even larger number of potential probe candidates. In addition, irreversible photoactivation can be achieved by "caging" fluorophores with a protective reactive moiety that is removed by irradiation with ultraviolet light. Unfortunately, however, there have been no reports to date of organic compounds or quantum dots that are capable of being photoconverted from one emission wavelength band to another.
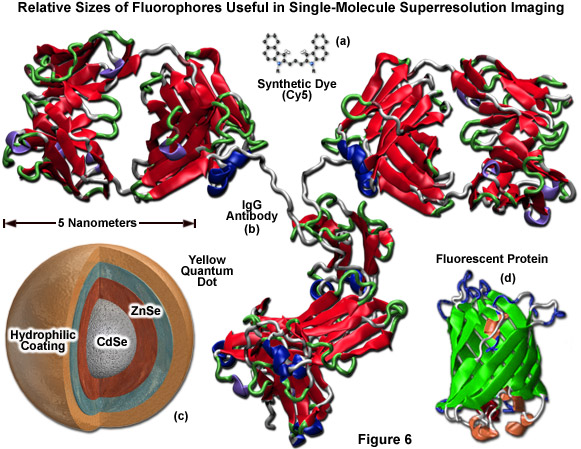
Illustrated in Figure 6 are the ribbon structures, molecular models, and/or cartoon drawings of the various fluorescent probes that are potentially useful in single-molecule superresolution imaging. The synthetic carbocyanine dye, Cy5 (Figure 6(a)) is significantly smaller than a yellow quantum dot (Figure 6(c)) or the typical beta-barrel can-shaped polypeptide architecture of a fluorescent protein (green ribbon structure; Figure 6(d)). Likewise, all of these probes are reduced in size compared to a primary or secondary IgG antibody, which can span 12 to 15 nanometers across (red ribbon structure; Figure 6(b)). Quantum dots must be treated with antibody fragments, streptavidin, phalloidin, or some other functional moiety in order to target specific sub-cellular regions. In many cases, the combined size of a quantum dot conjugated to one or more secondary antibodies can range up to 25 to 30 nanometers, which dramatically exceeds the size of a monomeric fluorescent protein. The smallest fluorescent probes are the synthetic dyes (Figure 6(a)), but these feature relatively poor targeting efficiency and generally generate large background signal.
The primary advantage of using synthetic fluorophores and quantum dots over fluorescent proteins for superresolution imaging is their high intrinsic brightness, excellent photostability, good contrast, and greater fatigue resistance. For example, tdEos yields approximately 750 photons per molecule in contrast to the over 6000 photons typically observed for the photoswitchable fluorophore combination of Cy3 and Cy5. Furthermore, the carbocyanine dyes can undergo over 200 switching cycles before photobleaching. The primary disadvantage of using quantum dots and synthetic fluorophores is the difficulty in targeting specific locations and high background signal when compared to fluorescent proteins. The most reliable targeting strategy for fluorophores in this class is conjugation to a primary or secondary antibody, although several new synthetic dyes have been demonstrated to localize in specific organelles independently.
The downside of using antibodies and immunofluorescence techniques to label intracellular structures for superresolution imaging is that the proteins are too large to permeate membranes and are therefore only useful in fixed and permeabilized cells unless the target is displayed on the outer region of the plasma membrane. Labeling with antibodies is also relatively low in efficiency and adds 10 to 20 nanometers to the localization uncertainty between the label and target. In addition, conjugates of quantum dots to antibodies do not perform on par with analogous conjugates using synthetic dyes such as the Alexa Fluors and carbocyanines. In perspective, however, the use of antibodies to target synthetic probes for PALM and related techniques provides a much higher signal level than fluorescent proteins, which are more useful in live-cell imaging. Unfortunately, the oxygen scavengers and thiols necessary to produce photoswitching with popular synthetic dyes are incompatible with living cells, so until a workaround is developed, fluorescent proteins will be the probes of choice for live-cell superresolution microscopy.
Quantum dots are inorganic semiconductor nanocrystals composed of a cadmium selenide (CdSe) core surrounded by a zinc sulfide (ZnS) shell that exhibit fluorescent properties owing to confined exciton emission. A passivation layer and hydrophilic coating must be applied to quantum dots for biological applications, and they must also be conjugated to streptavidin or antibodies for targeting. The fluorescence emission profile of quantum dots is remarkably symmetrical and generally exhibits a large quantum yield, whereas their broad absorption profile enables them to be excited over an unusually wide wavelength range. The size of the CdSe core dictates the emission spectral profile, with smaller cores (ranging down to approximately 2 nanometers) emitting in the blue and cyan regions and larger cores (5 to 7 nanometers) emitting in the yellow and red wavelengths.
In general, the photostability for quantum dots dramatically exceeds that of all other known fluorophores, including synthetic fluorophores and fluorescent proteins, which creates a problem for stochastic superresolution imaging unless quantum dots can be converted into a photoswitchable state. Recently, investigators demonstrated that manganese doping of ZnSe quantum dots can be used to generate a species that can be reversibly photoswitched with high efficiency using light without the requirement for external activators or quenchers (effectors). Targeting remains a problem with quantum dots; however, continued advancements in quantum dot chemistry will undoubtedly lead to new and better probes in this class.
Among the synthetic reversibly photoswitchable probes that have found utility in superresolution imaging are rhodamine derivatives, carbocyanine dyes (Cy2 through Cy7), Alexa Fluors, and ATTO dyes, with new candidates being developed and tested on a continuous basis. The photoswitchable cyanine dyes have been the most extensively used synthetic organic probes in stochastic superresolution imaging. The near-infrared dye, Cy5, has seen the most duty and can be used without an effector, although combining Cy5 with a secondary chromophore (such as Cy3, Cy2, or Alexa Fluor 405) dramatically facilitates photoswitching. As an example application, combining Cy5 with Cy3 enables the use of a red laser (635 nanometers) to photoswitch Cy5 to a stable dark state, while exposure to 543-nanometer light converts Cy5 back to the fluorescent species. The rate of conversion back to the fluorescent species depends on the proximity of the secondary effector (Cy3).
Additional combinations of cyanine and Alexa Fluor dyes, such as Cy3 and Cy5.5 or Cy7 and Alexa Fluor 647 with Cy2 or Cy3, have been reported, greatly expanding the available color palette for superresolution imaging. Furthermore, a large number of commercially available synthetics, including most of the Alexa Fluor and ATTO dye families, have been demonstrated to reversibly photoswitch in fixed cells using a protocol that includes thiol-containing reducing agents (β-mercaptoethylamine, dithiothreitol, or glutathione) to generate a stable non-fluorescent state. Thus, the potential for generating synthetic organic photoswitchers for multi-color imaging, even though the switching mechanism has yet to be determined, currently surpasses the limited palette of FPs that undergo light-induced modulation. Presented in Figure 7 are the structures of several synthetic probes illustrating the wide diversity of fluorophores that have been found useful for superresolution imaging.
Similar to the photoactivatable FPs (PA-GFP and PA-mCherry), a large library of caged synthetics would be especially useful for stochastic superresolution imaging. Unfortunately, only a few promising candidates have been reported to date: caged versions of fluorescein and rhodamine, which have been demonstrated to act in a manner similar to PA-GFP in PALM imaging. In practice, the caged synthetics are liberated from their protective ester groups using irradiation with ultraviolet light to generate a fluorescent species exhibiting excellent contrast that can be localized with high precision and then photobleached. Until a larger variety of caged fluorophores emerges, this class will remain limited in superresolution imaging applications.
The future of superresolution imaging relies on increasing the specificity and labeling efficiency of very bright photoswitchable fluorophores while simultaneously decreasing the size of the targeting peptides. A wide spectrum of hybrid systems designed to couple synthetic fluorophores with genetically-encoded targets may one day be capable of achieving this goal. All of these systems utilize a peptide or protein sequence that is expressed in living cells and is capable of recruiting a small synthetic molecule to bestow fluorescence. The most developed candidate in this class utilizes a tetracysteine motif fused to a variety of genetic targets to recruit blue, green, or red fluorophores (CHoAsH, FlAsH, and ReAsH, respectively) capable of binding to the cysteine residues to generate a probe similar in specificity to FPs. The major disadvantage of these combinations is the inability to overcome the high background levels of unbound fluorophore that lower contrast. A number of other hybrid system candidates have been developed, but none have seen significant use in superresolution imaging.
back to top ^Two-Color Superresolution Microscopy
Performing two-color single-molecule superresolution imaging requires a pair of fluorescent probes with emission spectra that are well separated and, in the ideal case, are photoswitched or photoactivated by illumination at different wavelengths. Currently, the palette of fluorescent proteins that are potentially useful for dual-color imaging is limited by emission spectral overlap conflicts, low photon output in probes having strategic spectral profiles (red emission), and the fact that most probes in this class are activated by light in the same wavelength region (ultraviolet or violet). There is significantly more potential for coupling two or more fluorophores together for simultaneous or sequential imaging in the growing class of synthetic probes, including the carbocyanines, Alexa Fluors, and ATTO dyes, which can be activated by laser lines ranging in wavelength from 405 to more than 647 nanometers. As the key to dual or triple color imaging in PALM and related techniques is the ability to identify and temporally separate the emission of each activated probe, the versatility afforded by synthetic dyes to use either emission, excitation, or activation wavelengths to distinguish between probes is a powerful asset.
The application of fluorescent proteins for multicolor imaging is rendered more difficult by the fact that most of the currently available red-emitting probes have pre-activation fluorescence emission that overlaps with the post-activation emission of green-emitting probes. For example, coupling Dronpa and tdEos together is hampered by the fact that the green spectral profile of Dronpa significantly overlaps the pre-converted profile of tdEos. However, investigators managed to image this pair of fluorescent protein in two-color PALM by photoactivating and imaging tdEos until all molecules were detected, localized, and photobleached before deactivating the Dronpa molecules using intense 488-nanometer light. Dronpa molecules were subsequently reactivated, localized, and combined with the tdEos images to construct a final two-color image. In general practice, however, depleting one fluorescent probe before imaging the other does not enable two-color imaging of the same region at multiple time points and has little potential for recording dynamic events.
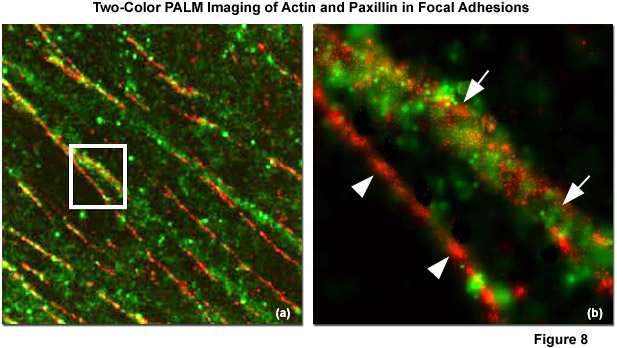
Illustrated in Figure 8 is the nanostructural organization of cytoskeletal actin and the focal adhesion protein, paxillin, in a human fibroblast cell (HFF-1 line) imaged with dual-color PALM. Figure 8(a) is a composite (dual color overlay) image of actin fused to the fluorescent protein Dronpa (pseudocolored green) and paxillin fused to the optical highlighter, tdEos (pseudocolored red), as they appear in a focal adhesion at high magnification. The white box in Figure 8(a) is enlarged in Figure 8(b) to reveal that there is very little direct overlap observed between actin and paxillin (white arrowheads), although actin bundles densely cluster around several fibrillar paxillin adhesions (white arrows). In widefield TIRF imaging, actin and paxillin appear to co-localize in focal adhesions, contrary to the information afforded by superresolution microscopy.
A superior approach for conducting two-color PALM experiments utilizes green and red emitting fluorescent proteins that have little or no spectral overlap. The development of a high photon output red-fluorescent photoactivatable mCherry fluorescent protein derivative, PA-mCherry1, has enabled dual color imaging with the green-fluorescent PA-GFP derivative of green fluorescent protein. Thus, PA-GFP and PA-mCherry1 are both switched on simultaneously with a 405-nanometer laser, while single-molecule fluorescence is imaged using 488-nanometer and 651-nanometer lasers, respectively. Although PA-mCherry1 will benefit from further improvements in performance, the probe can also potentially be used with several other green photoactivatable fluorescent proteins, such as Dronpa, rsFastLime, and PS-CFP2 to yield similar results. As new optical highlighters with improved photon output and spectral properties are developed, there will likely be dramatic improvements in the ability to conduct two-color PALM experiments with fluorescent proteins in both fixed and living cells.
The photoswitchable carbocyanine dyes offer a broad palette of probes for multicolor PALM and STORM imaging when coupled to activator fluorophores having varying activation wavelengths. The far-red and near-infrared dyes Cy5, Cy5.5, and Cy7, can function as photoswitchable acceptor reporters for a wide range of activator dyes that include Alexa Fluor 405, Cy2, and Cy3. Mixing the pairing of activators and reporters in this class expands the number of potential combinations that can be discriminated according to their activation or emission wavelengths for multicolor imaging. Furthermore, the carbocyanines Cy5 through Cy7 can also be spontaneously activated and deactivated with a single laser without the requirement for activators while still producing easily separated fluorescence emission spectra. A number of xanthene dyes, such as Alexa Fluors in the emission range between 500 and 647 nanometers and certain photochromic rhodamine dyes, as well as the ATTO dyes, can be photoswitched with the same laser, potentially enabling multicolor imaging. Fluorescent proteins can also be combined with synthetic dyes for multicolor imaging, as demonstrated for PALMIRA using rsFastLime paired with Cy5. As new probes are developed and tested for their utility in single-molecule superresolution imaging, the list of candidates for multicolor experiments will no doubt increase in the upcoming years.
back to top ^Three-Dimensional PALM Imaging
As originally developed, PALM and related single-molecule localization superresolution techniques were largely confined to imaging in the lateral, two-dimensional plane to acquire positional information about individual fluorophores. Probing the axial dimension was restricted due to the application of total internal reflection microscopy to implement imaging, and because specimens consisted of relatively thin sections produced with a microtome. Both methodologies confine imaging in the z plane to a depth of approximately 100 nanometers, which affords some latitude with respect to axial imaging of small superstructures, but is beneath the size of many others. Expanded vertical (axial) positional information can be revealed through a variety of techniques combined with photoactivation and localization. These methods can be grouped into several different categories, including exploitation of the z-dependent point-spread function, examining the specimen from a side-view approach, and employing interferometry along the axial direction.
In terms of an axial (z)-dependent point-spread function, the simplest example is defocusing of a point object (such as a single molecule emitter) that is positioned above or below the ideal in-focus lateral image plane. The image of such a point source can broaden, develop concentric rings around a central peak, or otherwise evolve into a form that deviates from the normal point-spread function. The basic approach to this type of imaging is to capture two simultaneous images at two different lateral planes, one underfocused and the other overfocused. This can be accomplished using a beamsplitter coupled with detectors set to image two different focal distances. The ratio of spot sizes is a monotonic function of axial position and can be easily calculated. Known as the biplane method, this approach has achieved within 20 to 30 percent of the standard PALM lateral resolution to yield a full-width at half maximum (FWHM) point-spread function of approximately 75 nanometers with fluorescent proteins. The primary advantage of biplane imaging is the relative simplicity in accessing the extra dimension.
A similar overfocus and underfocus approach can be implemented along two orthogonal axes of a single image, so that the x axis can focus at a different axial position than the y axis. The technique was first demonstrated with STORM through the addition of a weak cylindrical lens between the objective and the EMCCD detector. In practice, fluorescence from a single molecule produces an elliptical image featuring a ratio of x and y diameters that reveal the relative axial position of the emitter. Readout of fluorescent ellipticities has given approximately 20-nanometer lateral and 50 to 60-nanometer vertical resolution using carbocyanine (Cy3 and Cy5) dye pairs in STORM imaging. More exotic axial-dependent point-spread functions have been produced with a spatial phase modulator. As an example, a double point image can be generated from a single point source where the angular orientation of this doublet is dependent on the axial position. Thus, the angular orientation can be used as a readout of the axial coordinates.
As an alternative to spatial focusing, femtosecond pulses of excitation light can be focused in time, but a significantly more sophisticated microscope configuration is required and the technique is dependent on non-linear two-photon rather than single-photon linear excitation. The primary advantage of this method is that spurious excitation is reduced in regions removed from the area of interest. As such, this preserves photoactivatable fluorescence, enabling multiple 1-micrometer planes to form stacks of PALM images up to around 10-micrometers thick. A different strategy of three-dimensional imaging is to capture a PALM image from an orthogonal direction by using a side-view virtual image generated by a reflected mirror surface. The microscope setup involves mounting the specimen in a holder containing v-groove mirrors or simply on a strongly sloped mirror surface. Specialized software is required for calibration and to calculate data from the combined data.
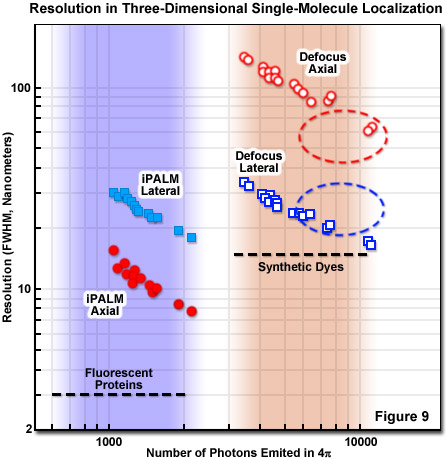
Perhaps the best resolution in three-dimensional single-molecule imaging is achieved with interferometry combined with PALM. The technique is known as interference-PALM (iPALM), and has demonstrated 10-nanometer vertical combined with 20-nanometer lateral localization precision using fluorescent protein labels. Interferometry is a common technique for measuring positions and is based on how light interferes after it has taken two different position-dependent pathways before being recombined with a beamsplitter. The special requirements of coherence, calibration, and tolerance of the highly unstable fluctuating nature of fluorescence require careful attention to the details of instrument configuration, which is best implemented with opposed objectives similar to a 4Pi system. Coherence can be maintained by realizing that any emitted photon is always coherent against itself, a critical concept for iPALM. Interference involving the same photon that takes two pathways with different axial-dependent path lengths satisfies this requirement. The self-calibration requirement is also fulfilled by single-photon self-interference and can be tolerant to fluctuations of a fluorescent source by using simultaneous multiphase interferometry provided by a specialized beamsplitter.
Presented in Figure 9 is a comparison of three-dimensional single-molecule localization techniques for fluorescent proteins and synthetic dyes with respect to the number of photons emitted and the axial and lateral resolution FWHM. The three-dimensional precision of iPALM and defocusing technique is shown as a function of the number of photons emitted from gold beads. iPALM (represented by filled circles and squares) exhibits better localization precision per emitted photon than defocusing techniques (open circles and squares) and displays better axial (filled red circles) than lateral (filled blue squares) resolution. In contrast, defocusing techniques have significantly less precision per emitted photon in the axial dimension (unfilled red circles) compared with the lateral direction (unfilled blue squares). Generally, most fluorescence microscopy techniques, including those for superresolution, exhibit poorer axial than lateral resolution, with iPALM being an exception.
For a complete evaluation of the different three-dimensional PALM approaches for a particular experiment, the practical constraints on sample parameters must be considered, including tolerable thickness, lateral size, timescale, probe labeling density and accuracy, along with resolution. It should be borne in mind that resolution is not just an instrumental number, but is in fact critically intertwined with the labeling approach. For example, brighter labels that produce more photons yield better localization accuracy but are tempered by considerations such as antibody linkage lengths, targeting specificity, and the density of molecular labeling. Endogenous fluorescent protein labels, such as tdEos, have reduced brightness and require a more efficient localization procedure to acquire satisfactory data. The trade-offs involved for iPALM imaging and defocusing techniques are presented in Figure 9.
back to top ^Live-Cell Imaging with PALM
The repetitive process of photoactivation, imaging, and photobleaching involved with PALM and similar single-molecule localization techniques imposes significant temporal constraints on observing specimens. In order to migrate the technique to live-cell imaging, the PALM process can be expedited by increasing both activation and excitation light intensities to hasten the turnover of molecules, but the living cells must be carefully monitored to ensure they are healthy. Although live-cell imaging with superresolution microscopy is still in its infancy, several investigations serve to fortify the foundation upon which future research will likely be based. These include using optical highlighter fluorescent proteins to create time-lapse PALM images, to determine the average distributions or distances between molecules in living cells, and to create dynamic molecular maps. Similar to the techniques described above, the imaged molecular density in any single frame of a time-lapse PALM experiment must remain low enough to isolate single molecules.
An important point to note is that in live-cell PALM, the density of localized molecules must be high enough to create a superresolution image if the aim is to measure the motions and rearrangements of protein-enriched structures in sub-diffraction precision. For this reason, slow-moving macromolecular structures, such as focal adhesion complexes, the filamentous actin cytoskeleton, microtubules, intermediate filaments, or endoplasmic reticulum morphological changes are appropriate candidates. In practice, approximately 500 to 1000 molecules per frame must be imaged as quickly as possible with the instrument configuration (data collection intervals should be restricted to a minute or less). Live-cell imaging with PALM has been conducted on focal adhesions to reveal that an individual adhesion complex moved at variable rates, recruited or lost different overall numbers of molecules, and underwent different shape changes. Future studies will address a host of other imaging scenarios.
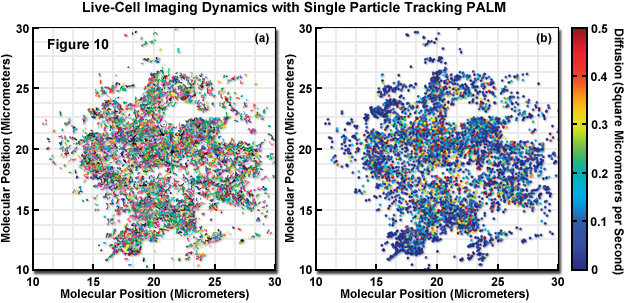
In a unique implementation of live-cell PALM imaging (see Figure 10), thousands of individual fluorescent protein fusions were tracked with a technique known as single-particle-tracking PALM (sptPALM). The trajectories of the fluorescent proteins were used to compare the distribution and dynamics of two viral proteins (ts045 vesicular stomatitis viral G protein, VSVG; and the human immunodeficiency virus type 1 structural protein, Gag) expressed in the plasma membrane. In Figure 10, the molecular motion of VSVG-tdEos fluorescent protein was imaged in a COS7 cell using 561-nanometer light while simultaneously photoactivating subsets of the tdEos molecules. The molecules were localized in each frame and their determined positions in consecutive frames were linked into tracks. The tracks in Figure 10(a) represent molecules that remained fluorescent for more than 0.75 seconds (plotted as different colors to distinguish individual tracks). The diffusion coefficient for each track was determined and plotted as a filled circle at the start of the track (Figure 10(b)). Each track has been assigned a color based on its diffusion value.
The result of sptPALM imaging in Figure 10 was spatially resolved maps of single-molecule diffusion coefficients, which revealed strikingly different diffusional behavior. VSVG molecules were determined to be highly mobile to freely explore the plasma membrane, whereas Gag proteins were often found in immobile clusters approximating the size of virus-like particles between 100 and 200 nanometers in diameter. Similar experiments in bacteria using Dendra2 tracked the mammalian tubulin homolog, Ftx, which forms a helical filamentous structure (termed a Z-ring) that is involved in bacterial cytokinesis. In contrast to superresolution videos of focal adhesions that capture slow dynamics, molecular single particle tracking can reveal far more rapid dynamics in ensembles of molecules.
Conclusions
Although still an emerging technique, single-molecule superresolution microscopy is receiving increasing attention among biologists interested in understanding cellular processes at the nanoscale. A number of methodologies are currently available and more are being developed to circumvent the diffraction-limited resolution barrier imposed by widefield and confocal fluorescence microscopy. Illumination-based superresolution techniques, such as STED, GSD, and SSIM, have pushed resolutions into the 30 to 50 nanometer range using conventional fluorophores, whereas single-molecule localization techniques (PALM and STORM) require special photoactivatable probes. Conventional probes have been successfully used in single-molecule localization by relying on transitions of molecules between light and dark states, but these may well come with the loss of molecular density during pre-experiment irradiation steps necessary to lower the fluorescence to single-molecule imaging levels.
The primary driving force behind PALM is the utter simplicity in both concept and instrumentation, requiring only a modified widefield fluorescence microscope (to conduct single-molecule imaging) and the ability to express fluorescent fusions in adherent cell cultures. However, the future of PALM and related single-molecule localization techniques will likely be enlightened with more advanced instrumentation and new fluorophores that are better photon emitters with excellent photoswitching characteristics, and exhibit fluorescence over the entire visible spectrum. Also, researchers must be mindful that these techniques began in earnest less than a decade ago and can still be considered in the developmental stage. Regardless, the techniques are sufficiently well developed that commercial instrumentation is now available. In the future, refinements in the photophysical behavior of fluorescent probes coupled to improvements in instrumentation and analysis could enhance the speed of experiments and significantly decrease acquisition times. Despite these growing pains, the rapid pace of superresolution microscopy gives confidence that imaging below the diffraction barrier will, in the next several years, become as commonplace as confocal imaging is today.
Contributing Authors
Eric Betzig and Harald F. Hess - Howard Hughes Medical Institute, Janelia Farm Research Campus, Ashburn, Virginia, 20147.
Hari Shroff - Section on High Resolution Optical Imaging, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, Bethesda, Maryland, 20892.
George H. Patterson - Biophotonics Section, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, Bethesda, Maryland, 20892.
Jennifer Lippincott-Schwartz - Cell Biology and Metabolism Program, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland, 20892.
Michael W. Davidson - National High Magnetic Field Laboratory, 1800 East Paul Dirac Dr., The Florida State University, Tallahassee, Florida, 32310.