
Introduction
The photophysical properties of fluorescent proteins are often extremely complex and can involve several distinct emissive and non-emissive (dark) states, as well as on-and-off blinking behavior when observed at the single molecule level. In fact, one of the first observations of photoswitching in fluorescent proteins was reported in wild-type GFP and several of its enhanced derivatives. As we will see, the photoswitching properties can allow the investigator to change the color or the emission state of a fluorescent protein, providing unique opportunities to track the dynamic behavior of proteins in living cells and animals. This quality can be extremely useful since, unlike the standard fluorescent proteins that are uniformly fluorescent from the time they are produced, photosensitive fluorescent proteins can be switched on at a particular time and location within the cell to track the behavior of a tagged protein. Although fluorescent proteins are known to undergo a variety of light-induced switching characteristics, the most useful are photoactivation, photoconversion, and photoswitching, functions that are collectively termed optical highlighting. Photoactivatable fluorescent proteins are capable of being activated from very low level to bright fluorescence emission upon illumination with ultraviolet or violet light, whereas photoconvertable fluorescent proteins can be optically converted from one fluorescence emission bandwidth to another. In contrast, photoswitchable fluorescent proteins have emission characteristics that can alternatively be turned on or off with specific illumination.
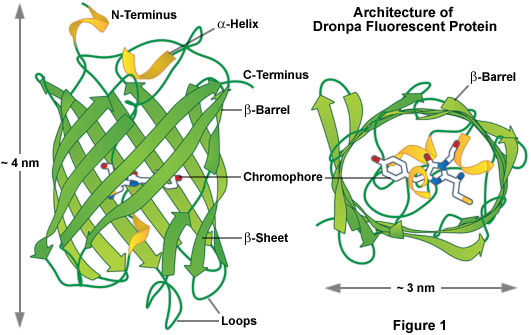
As a class, optical highlighters, such as the photoswitchable Dronpa illustrated in Figure 1, allow direct and controlled activation of distinct molecular pools of the fluorescent proteins within the cell. The investigator selects the population of photoactivated or photoconverted molecules to be followed, so dynamic behavior can be monitored over time, independent of other newly synthesized proteins. An ideal optical highlighter protein should be readily photoconvertable (through the process of fluorescence activation and/or emission wavelength shifts) to produce a high level of contrast, as well as being monomeric for optimum expression in the target system. These probes are especially useful since measurements of the photoactivated or photoconverted populations of proteins are not influenced by newly synthesized or non-converted proteins, which either remain invisible or continue to emit the original wavelengths. In addition, the time required for photoactivation is typically very brief (often less than a second), which allows direct investigations of extremely rapid cellular processes. By repeated excitation in the region of interest, optical highlighters can be continuously photoconverted at a specific intracellular location. Optical highlighters are also emerging as important tools in superresolution microscopy where their switching properties can be utilized to selectively convert limited numbers of molecules in a much larger pool.
Photoactivatable Fluorescent Proteins
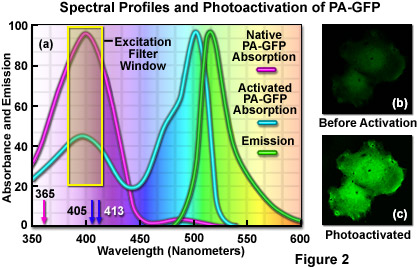
The unique photophysical properties of wild-type green fluorescent protein (wild-type GFP) were thoroughly investigated during the mid-1990s, and served as a foundation for the creation of the first useful optical highlighter designed specifically for photoactivation studies. Termed PA-GFP (for Photo Activatable Green Fluorescent Protein), this optical highlighter was developed by improving the photoconversion efficiency of the native chromophore from a predominately neutral form to a species that is anionic in character. By replacing the threonine at position 203 with a histidine residue (T203H) in wild-type GFP, researchers produced a variant having negligible absorbance in the region between 450 and 550 nanometers, thus dramatically enhancing contrast between the non-activated and activated species (see Figure 2). PA-GFP is optimally excited at 400 nanometers, but has negligible absorbance in the region between 450 and 550 nanometers. However, after photoactivation with violet light, the absorption maximum of PA-GFP is shifted to 504 nanometers, increasing green fluorescence when excited at 488 nanometers by approximately 100-fold and providing very high contrast differences between the converted and unconverted pools. When PA-GFP-labeled proteins are photoactivated inside the living cell, the diffusion of the newly fluorescent proteins provides a direct measure of the mobility of the labeled proteins. The major drawback in the use of PA-GFP is that the non-activated form is not readily distinguishable before photoactivation, making it difficult to identify the regions that are expressing the fluorescent protein.
An optical highlighter derived from aceGFP, isolated from Aequorea coerulescens, has been reported to transition from cyan (468 nanometers) to green fluorescence (511 nanometers) upon illumination at 405 nanometers. Named PS-CFP (PhotoSwitchable Cyan Fluorescent Protein), this unique highlighter is particularly advantageous because a significant level of cyan fluorescence is present prior to photoconversion, a feature that enables investigators to track and selectively illuminate specific intracellular regions. Unfortunately, the fluorescence emission intensity of PS-CFP (and its commercially available version, PS-CFP2; Evrogen) is approximately 2-fold less than PA-GFP, significantly reducing the contrast ratio. The photoactivation mechanism of both PA-GFP and PS-CFP (see Figure 3) is believed to be light-induced decarboxylation of the glutamic acid side chain in residue 222 (Glu222), which shifts the chromophore equilibrium to the anionic form.
Several photoactivatable optical highlighters have been produced using site-directed mutagenesis of monomeric red-shifted reef coral fluorescent proteins. The first variant, derived from mRFP1, was named PA-mRFP1, and it exhibits a 70-fold increase in fluorescence intensity upon activation using ultraviolet or short wavelength violet illumination. However, the relatively low level of fluorescence observed with PA-mRFP1 (approximately 7 percent of PA-GFP) has prevented the widespread use of this highlighter. Shifting the focus to more advanced red fluorescent proteins, investigators recently reported a photoactivatable variant of mCherry (PA-mCherry1) having excitation and emission spectra at 564 and 595 nanometers, respectively. Compared to PA-mRFP1 the mCherry version features faster maturation, better pH stability, faster photoactivation, improved photostability, and higher contrast. This highlighter should be an excellent complement to PA-GFP for dual color photoactivation labeling in living cells and for superresolution microscopy investigations.
PA-GFP serves as the activation agent in a new optical highlighter design based on FRET. Termed Phamret (an acronym for Photoactivation-mediated resonance energy transfer), this unique probe couples PA-GFP to a high-performance ECFP variant through a two amino acid linker. When excited with 458-nanometer light, Phamret fluoresces cyan so that fusion with targeting peptides or proteins can be readily identified and selected for photoconversion. The PA-GFP portion of Phamret can then be photoactivated with 405-nanometer illumination to evoke FRET between the ECFP moiety and activated PA-GFP. After photoactivation, Phamret exhibits green fluorescence (520 nanometer peak) upon illumination at 458 nanometers and can therefore be used as an optical highlighter. One of the advantages of Phamret is that imaging can be conducted using only a single excitation wavelength for both the native and photoactivated species. The major downside of Phamret is that two fluorescent protein units were used to construct the probe (similar to a tandem dimer), increasing the size and possibly creating steric hindrance in some fusions.
Although researchers have made significant progress in the development of photoactivatable probes, there remains much room for improvement. The photoactivatable optical highlighter color palette is still very limited, and these probes are difficult to track in the non-activated form. Furthermore, the brightness of the photoactivated species in all reported variants is less than 50 percent that of EGFP and their photostability is significantly diminished compared to the parent fluorescent proteins. Continued progress in this area likely will yield an expanded color palette of photostable optical highlighters that are useful in a variety of studies.
Photoconvertable Fluorescent Proteins
One of the most useful classes of optical highlighters encompasses the growing number of fluorescent proteins reported to undergo photoconversion from one emission wavelength to another. Unlike photoactivatable fluorescent proteins, these probes are readily tracked and imaged in their native emission state prior to photoconversion, making it easier to identify and select regions for optical highlighting. The first report of a photoconvertable highlighter was a tetrameric fluorescent protein isolated from the stony Open Brain coral, Trachyphyllia geoffroyi, which can be photoconverted from green to red fluorescence emission by illumination with ultraviolet light. The discovery of this highlighter was serendipitous, as are many important discoveries. It occurred when the researchers accidentally left a test tube containing the protein on a laboratory bench near a window, and then astutely observed the shift from green to red. The unusual color transition prompted investigators to name the protein Kaede, after the leaves of the Japanese maple tree that turn from green to red in the fall.
Illumination of the commercially available (MBL International) Kaede optical highlighter between 380 and 400 nanometers results in a rapid spectral shift from the principal maxima at 508 nanometers (absorption) and 518 nanometers (emission) to longer wavelength peaks at 572 and 582 nanometers, respectively. Along with photoconversion, there is a dramatic increase in the red-to-green fluorescence ratio (approximately 2000-fold, considering both the decrease in green and the increase in red emission). Photoconversion in Kaede is stable and irreversible under aerobic conditions. Neither exposure to dark for extended periods, nor strong illumination at 570 nanometers, can restore green fluorescence to the chromophore. The red fluorescent state of the Kaede chromophore is comparable to the green in brightness and stability, and because the unconverted protein emits very little fluorescence above 550 nanometers, the appearance of strong red signal provides excellent contrast. The major drawback of Kaede is the tetrameric nature of the protein, which limits its use in most fusion applications in live cell imaging, although the highlighter has been widely reported to be useful in transgenic studies.
Properties of Selected Optical Highlighter Fluorescent Protein Variants
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 1
A compilation of properties of the most useful optical highlighter fluorescent protein variants is presented in Table 1. Along with the common name and/or acronym for each highlighter, the peak excitation (Ex) and emission (Em) wavelengths, molar extinction coefficient (EC), quantum yield (QY), relative brightness, photostability, and physiologically relevant quaternary structure are listed. The computed brightness values were derived from the product of the molar extinction coefficient and quantum yield, divided by the value for EGFP (ND* indicates the values have not been determined). This listing was created from scientific and commercial literature resources and is not intended to be comprehensive, but instead represents fluorescent protein derivatives that have received considerable attention in the literature and may prove valuable in research efforts. The excitation and emission peak values listed may vary in published reports due to the broad spectral profiles. In actual fluorescence microscopy investigations, the experimental brightness of a particular fluorescent protein may differ (in relative terms) from the brightness provided in this table. Among the many potential reasons for these differences are wavelength-dependent variations in the transmission or reflectance of microscope optics and the efficiency of the camera. Furthermore, the extent of fluorescent protein folding and maturation will depend on both the particular variant being used as well as the characteristics and localization of the fusion partner.
Similar fluorescent proteins capable of being photoconverted from green to red fluorescence emission by violet and ultraviolet illumination have been discovered in the Great Star coral (mcavRFP; derived from Montastraea cavernosa), soft corals (DendFP; derived from members of the genus Dendronephthya), and the mushroom coral (rfloRFP; derived from Ricordea florida). Each of these optical highlighters (including Eos and KikGR, discussed below) contain a chromophore derived from the tripeptide His62-Tyr63-Gly64 that initially emits green fluorescence until driven into a red state by a light-catalyzed cleavage of the polypeptide backbone. These amino acids form the imidazolinone chromophore (similar to the Aequorea proteins). Irradiation induces cleavage between the amide nitrogen and alpha-carbon atoms in the histidine residue with subsequent formation of a highly conjugated dual imidazole ring system, a process requiring catalysis by the intact protein and resulting in the dramatic shift of fluorescence emission to red wavelengths. The unconventional chemistry involved in this chromophore transition should provide fluorescent protein engineers with an excellent foundation upon which to develop more advanced highlighters. A monomeric variant of the tetrameric DendFP has been developed through random and site-directed mutagenesis and named Dendra (from Dendronephthya and red activatable). This highlighter features excitation and emission maxima for the green and red forms of 490/507 nanometers and 553/573 nanometers, respectively, and functions well in fusion tags for subcellular localization. The commercial version, Dendra2 (Evrogen) is the first monomeric red-to-green optical highlighter that has enjoyed widespread use as a tracking tool in live cell imaging.
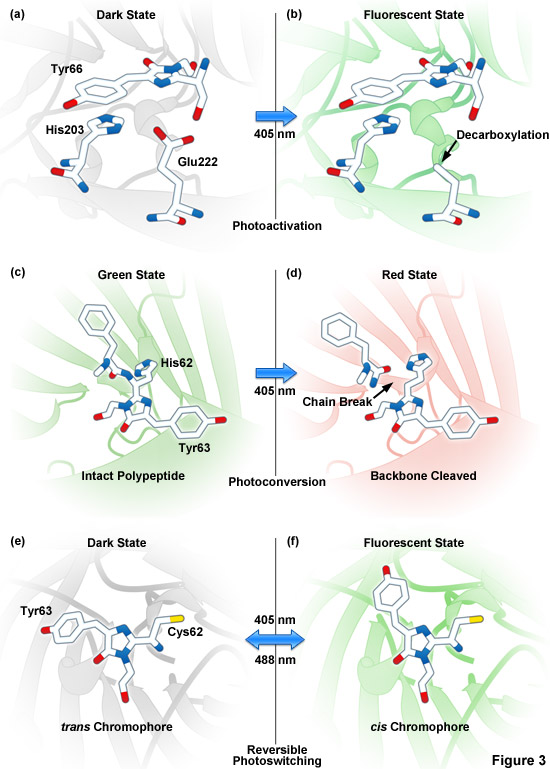
Presented in Figure 3 are photoactivation, photoconversion and photoswitching mechanisms for optical highlighter fluorescent proteins. Figure 3(a) and Figure 3(b) illustrate photoactivation of PA-GFP and PS-CFP2, which is believed to occur due to decarboxylation of Glu222 (or the equivalent) followed by conversion of the chromophore from a neutral to anionic state. Green-to-red photoconversion for Kaede, KikGR, Dendra2, and Eos is depicted in Figures 3(c) and 3(d). These optical highlighters all contain the HYG chromophore, and photoconversion occurs when the fluorescent protein is illuminated with ultraviolet or violet radiation (405 nanometers) to induce cleavage between the amide nitrogen and alpha-carbon atoms in the histidine 62 residue leading to subsequent formation of a conjugated dual imidazole ring system. Photoswitching of Dronpa (Figures 3(e) and 3(f)) involves cis-trans photoisomerization induced by alternating radiation between 405 nanometers and 488 nanometers. A similar isomerization mechanism is suggested to operate in mTFP0.7 and KFP1.
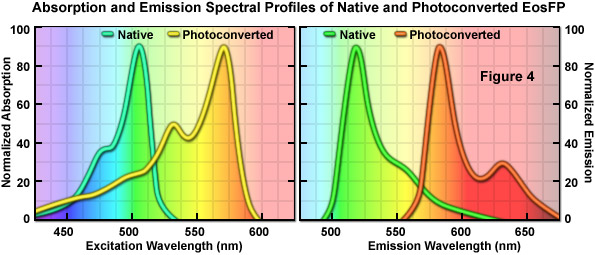
An optical highlighter isolated from another stony coral (Lobophyllia hemprichii), the tetrameric EosFP (named after the goddess of dawn in Greek mythology), behaves similarly to Kaede and the other variants described above. EosFP emits bright green fluorescence at 516 nanometers, and can be photoconverted to orange-red (581 nanometers) fluorescence when illuminated at near-ultraviolet wavelengths (see Figure 4). Random and site-directed mutagenesis of tetrameric EosFP was used to generate two dimers and a true monomeric protein named mEosFP. Unfortunately, the monomeric variant of EosFP can only be expressed efficiently at temperatures below 30° C, limiting the utility of mEosFP in mammalian systems. To create a pseudo-monomer suitable for imaging fusions at 37° C, investigators linked two of the dimeric EosFP units together using a 16-amino acid linker to produce a tandem dimer. tdEosFP has turned out to be one of the most useful optical highlighters yet developed because of its high functionality as a tag in fusion vectors and for superresolution imaging. Recently, an improved monomeric version that matures at 37° C, named mEos2, has been reported. Although not as bright as tdEos, the mEos2 derivative is an excellent complement for imaging problematic fusions (tubulin, histones, gap junctions) that localize poorly with tandem dimers.
A third stony coral, Favia favus, has yielded a promising tetrameric fluorescent protein that exhibits efficient photoconversion from green to red fluorescence emission wavelengths (similar to Kaede) upon irradiation with near-ultraviolet or violet light. Genetic engineering efforts based on structural analysis of this protein produced a tetrameric variant, termed KikGR (named after Kikume-ishi, the Japanese term for Favia), which is several-fold brighter than Kaede in both the green and red states when expressed in mammalian cells. Commercially available (MBL International) under the name Kikume Green-Red, the highlighter has been demonstrated to be successfully photoconverted using multiphoton excitation at 760 nanometers, which can be used for specific labeling of cells with high spatial resolution in thick tissues. Furthermore, the KikGR highlighter features a wider separation of green and red emission maxima than Kaede (75 nanometers versus 54 nanometers, respectively). Mutagenesis of the tetrameric KikGR yielded a monomeric derivative containing 21 mutations, which is termed mKikGR and has been demonstrated to perform well in fusions that do not tolerate tetramers. Both the green and red form of mKikGR are less photostable and dimmer than mEos2, although mKikGR might perform better in tubulin and gap junction fusions.
Of the currently available monomeric green-to-red optical highlighters (mEos2, Dendra2, and mKikGR), the native and photoconverted species of mEos2 are the brightest followed by Dendra2 and mKikGR, respectively. Dendra2 and mEos2 are almost equivalent in photostability for both the green and red species, and far more photostable than mKikGR. All three of these optical highlighters should be useful in a wide variety of dynamic tracking experiments and can be used for precise localization in superresolution microscopy. Significantly, it appears that photoconversion in fluorescent proteins is apparently more widespread than originally suspected as evidenced by recent observations of alterations to the emission spectral profiles of cyan and yellow fluorescent proteins following photobleaching experiments. Researchers have discovered that several of the orange and red Anthozoa proteins, including mOrange, mKate, and HcRed1, have been observed to shift emission to longer or shorter wavelengths upon intense illumination using single and two-photon laser sources, although the contrast ratios between the native and photoconverted species are low. Thus, photoconversion can be harnessed as a tool for cellular dynamics and superresolution microscopy, but investigators should also be aware that this phenomenon could surface as an artifact during photobleaching, FRET, or imaging experiments.
Photoswitchable Fluorescent Proteins
The phenomenon of photochromism (the ability to switch between fluorescent and dark states) has been observed in wild-type GFP and several yellow fluorescent protein derivatives at the single molecule level. However, none have demonstrated this phenomenon when measured in bulk. In single molecule studies, fluorescent proteins exhibited fluorescence for several seconds during illumination at 488 nanometers followed by an equally short interval without fluorescence, after which the fluorescence resumed. The on-and-off switching or blinking behavior was repeated many times before each fluorescent protein molecule ultimately photobleached. Unfortunately, photoswitching in most of the fluorescent proteins described above is stochastic, and cannot be used in quantitative experiments. However, some optical highlighters have been isolated that can be reliably toggled on or off by illumination with different excitation wavelengths, and these are called photoswitchable fluorescent proteins.
The most prominent and well-studied member of this class is named Dronpa (Figure 1; named after a fusion of the Ninja term for vanishing and photoactivation), which is a monomeric variant derived from a stony coral tetramer. Dronpa fluorescent protein exhibits an absorption maximum at 503 nanometers (arising from the anionic, deprotonated chromophore) with a minor peak at 390 nanometers (from the neutral, protonated chromophore). The anionic chromophore emits green fluorescence with a maximum at 518 nanometers and has a brightness level almost 2.5 times that of EGFP. Dronpa photoswitching occurs in part by interconversion between the deprotonated (on state; bright) and protonated (off state; dark) forms. Illumination at 488 nanometers drives Dronpa to the dark species after which the fluorescent protein can be subsequently switched back on by brief illumination at 405 nanometers. This cycle can be repeated several hundred times without significant photobleaching. The primary mechanism of fluorescent protein photoswitching is thought to also arise from cis-trans isomerization of the hydroxybenzilidine (tyrosyl side chain) chromophore moiety that accompanies the changes in the protonation state. Similar to other highlighters, Dronpa is useful both for dynamics and superresolution studies. Variants of Dronpa (see Table 1) with faster switching times, reversed photoswitching properties, and broader spectra have been reported, but application of these rather dim fluorescent proteins has so far been limited to evaluating their performance in superresolution imaging. Future versions will no doubt feature higher brightness and should prove useful in routine dynamics investigations.
Several photoswitching fluorescent proteins in other regions of the color palette have been developed from anemones and corals. Kindling fluorescent protein (commercially available from Evrogen as KFP1) is a tetrameric highlighter that emits red fluorescence at 600 nanometers upon illumination with green or yellow light (525 to 580 nanometers). Upon cessation of illumination, KFP1 relaxes back to its initial non-fluorescent state. Irradiation with intense blue light (450 to 490 nanometers) completely quenches KFP1 fluorescence immediately, enabling control over the photoswitching. A cyan fluorescent protein from coral named mTFP0.7 (an intermediate in mTFP1 mutagenesis) has also been demonstrated to photoswitch, but has not been characterized in living cells. In addition, monomeric photoswitchable variants of mCherry have recently been introduced as potential probes for superresolution microscopy. Termed rsCherry and rsCherryRev, these derivatives display antagonistic switching modes. Here, irradiation of rsCherry with yellow light induces the bright state and blue light drives the fluorescent protein to the dark state, whereas the reverse is observed with rsCherryRev. However, these fluorescent proteins are only approximately 10 percent as bright as mCherry when expressed as ensembles in cells, but are equally bright as mCherry on the single molecule level.
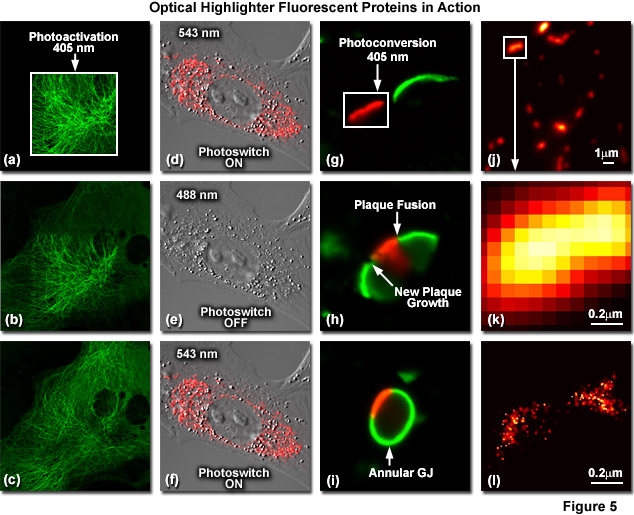
Illustrated in Figure 5 are optical highlighter fluorescent proteins in action imaged with laser scanning confocal microscopy. Figure 5(a) through 5(c) show photoactivation of mPA-GFP-tubulin in opossum kidney (OK cell line) epithelial cells. In Figure 5(a), a rectangular region of interest is illuminated at 405 nanometers for 5 seconds at time zero. The photoactivated tubulin chimera slowly migrates to other portions of the cell in Figure 5(b) at 25 minutes. The microtubule network subsequently gains more intensity after 1 hour (Figure 5(c)). Photoswitching of the mitochondria with a fusion of KFP1 to a mitochondria targeting sequence in fox lung fibroblast cells is followed in Figures 5(d) through 5(f). Labeled mitochondria are imaged with a 543-nanometer laser in both fluorescence and differential interference contrast in Figure 5(d). After completely photoswitching the KFP1-labeled chimera "off" with 488 nanometer illumination (Figure 5(e)), the mitochondria now appear devoid of fluorescence. The KFP1 label in mitochondria is reactivated with illumination at 543 nanometers (Figure 5(f)), and does not significantly photobleach after 5 rounds of photoswitching. Photoconversion of gap junctions labeled with a fusion of mEos2 to connexin Cx43 in HeLa cells is presented in Figures 5(g) through 5(i). Photoconversion of a gap junction plaque (red) in a selected region (white box) with 405-nanometer illumination at time zero is shown in Figure 5(g). New plaque growth and fusion of a non-converted plaque occurs after approximately 50 minutes (Figure 5(h)), followed by formation of an annular gap junction having a photoconverted region at 80 minutes (Figure 5(i)). Superresolution microscopy (PALM) imaging of tdEos-mitochondrial targeting sequence is shown Figures 5(j) through 5(l). A widefield total internal reflection fluorescence (TIRF) image of a mitochondria field near the nucleus is shown in Figure 5(j), along with a summed PALM image (Figure 5(k)) of boxed region in Figure 5(j) and a PALM image (Figure 5(i)) of the mitochondrial fusion.
A new and potentially very useful optical highlighter derived from wtEosFP couples the properties of photoconversion and photoswitching into a single reporter. Named IrisFP, a single mutation in wtEosFP (F173S) bestows reversible photoswitching induced by cis-trans isomerization of the chromophore in both the native (green) and photoconverted (red) species. Similar to the wild-type parent, IrisFP undergoes photoconversion from a green to a red-emitting state upon illumination with violet (405 nanometers) or ultraviolet light due to extension of the conjugated aromatic electron system that accompanies cleavage of the polypeptide backbone. Illumination of the green IrisFP species with 488-nanometer laser light drives the highlighter to a non-fluorescent dark state, which can revert to a bright species upon illumination with low-intensity 405-nanometer light. High intensity 405-nanometer illumination drives the IrisFP chromophore to the red state. This state can be photoswitched off with 532-nanometer light and back on again with 440-nanometer light. Although hampered for use in most fusion tags by the tetrameric quaternary structure, IrisFP represents a new class of optical highlighters that will perhaps be exploited in the future.
Investigations into the underlying mechanism of fluorescent protein photoswitching indicate that cis-trans isomerization of the tyrosine moiety in the chromophore is a key event in the process. The cis conformation represents the bright fluorescent state, whereas the trans conformation reverts the chromophore to the non-fluorescent dark state. These conformational changes are thought to be accompanied by varied chromophore protonation states that also contribute to the determination of the fluorescent properties. In addition, photoswitching is to some degree a manifestation of chromophore planarity and structural rearrangements of internal amino acid side chains within the chromophore cavity. These collective features may constitute a fundamental mechanism that is common to all photoactivatable and reversibly photoswitchable fluorescent protein derivatives.
Fluorescent Timer Proteins
A unique, but limited class of fluorescent proteins that change spectral properties with time are referred to as fluorescent timers, the first example of which was a variant of DsRed. Termed E5, this fluorescent protein derivative effectively functions as a fluorescent clock that slowly changes emission from green to red over a time period of several hours to provide temporal and spatial information on gene activity. Thus, green fluorescent regions indicate recent gene activation, while yellow-orange regions signify continuous activity. Areas exhibiting bright red fluorescence indicate that gene promoter activity has ceased for an extended period. Fluorescent protein timer E5 was used in vivo to demonstrate expression in Caenorhabditis elegans and Xenopus promoters. However, because E5 is an obligate tetramer, it has limited promise as a protein fusion tag.
A recent study focused on improving fluorescent timers yielded three monomeric variants based on mCherry that change fluorescence from blue to red over time. The mCherry derivatives are divided into distinct classes that exhibit fast, medium, and slow blue-to-red chromophore maturations rates that are temperature dependent (see Table 1). The fastest variant exhibits a shift of blue to red fluorescence in approximately 15 minutes, whereas the medium and slow variants have corresponding times of 1.2 and 9.8 hours, respectively. These fluorescent protein timers were demonstrated to be useful when expressed in bacteria, as well as insect and mammalian cells. Continued development of fluorescent timers should yield multi-colored variants with improved brightness and photostability for long-term experiments.
Conclusions
The optical highlighter fluorescent proteins produced thus far from Aequorea GFP derivatives and reef coral proteins show sufficient promise to warrant aggressive efforts to solve problems associated with oligomerization, limited brightness, and low photostability as well as fine-tuning of their spectral profiles. Further engineering to generate more advanced optical highlighters should shift photoactivation wavelengths to the less phototoxic blue and green spectral regions while simultaneously pushing emission wavelengths into the yellow through far-red regions to significantly expand the potential applications of these probes.
Contributing Authors
David W. Piston - Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville, Tennessee, 37232
Robert E. Campbell - Department of Chemistry, University of Alberta, Edmonton, Alberta, Canada, T6G2G2
Richard N. Day - Department of Cellular and Integrative Physiology, Indiana University School of Medicine, 635 Barnhill Dr., Indianapolis, Indiana, 46202.
Michael W. Davidson - National High Magnetic Field Laboratory, 1800 East Paul Dirac Dr., The Florida State University, Tallahassee, Florida, 32310.